الأعراض
تختلف أعراض المرض على حسب نوع الثلاسيميا المصاب بها الشخص.وقد لا يشعر المصاب بالمرض بأي مشكلة كما هو الحال فالشخص الحامل للألفا ثلاسيميا وتكتشف بالمصادفة عند إجراء تحليل دم لكريات الدم الحمراء والهيموجلوبين.و في المقابل قد يكون شديد كما هو الحال في البيتا ثلاسيميا الكبر في تكون الأعراض واضحة و تتفاقم المشكلة إذا لم ينقل للمريض دم بشكل دوري.كما أن المرض قد يكون شديد لدرجة أن الجنين يموت في بطن أمة أو يولد مع استسقاء في جميع الجسم نتيجة لنقص الهيموجلوبين في الدم وهبوط شديد في القلب،وهذا يحدث في الألفا ثلاسيميا التي يكون فيها جميع مورثات(جينات)الألفا هيموجلوبين معطوبة (بها طفرة مرضية).
ضعف في البنية مع انتفاخ في البطن مصحوب بتضخم في الكبد والطحال
وفيما يلي الأعراض التي تحدث للمصابين بمرض البيتا ثلاسيميا الكبرى إذا لم يعالج المريض:
.شحوب واصفرار البشرة والشفتين
الخمول و الشعور بالتعب والإرهاق لأقل جهد.
فقدان الشهية.
.نقص حاد في الهيموجلوبين (اقل من 9 مج\مل دم)
تضخم الكبد والطحال نتيجة عجز نخاع العظام عن إنتاج كريات الدم الحمراء فيبدأ الكبد والطحال وأعضاء أخرى بمحاولة تصنيع الدم بنفسها مما يؤدي إلى تضخمها.
الانعكاسات النفسية السيئة لدى المريض نتيجة شعوره بالمرض الدائم والنقص الصحي مقارنة بأقرانه.
التأخر في النمو الجسماني كالطول والوزن .
تغيرات في عظام الجسم ومنها الجمجمة ، بروز الجبهة وعظام الوجنتين ، انخفاض عظام الأنف وبروز عظام الفك العلوي.
زيادة الالتهابات بشكل عام.
سرعة ضربات القلب لمحاولة تعويض نقص الهيموجلوبين وذلك بزيادة سرعة ضخ القلب للدم، كذلك قد تحدث مضاعفات قلبيه مع مرور الوقت.
حدوث مضاعفات نتيجة تراكم الحديد في أعضاء مختلفة من الجسم أن لم ينتظم المريض باستعمال الحقنه اليومية للدسفيرال والتي تؤدي إلى التخلص من الحديد عن طريق البول وتتمثل هذه المضاعفات في تشحم الكبد ، اسوداد لون الجلد، خلل هرموني (مرض السكري) ، انخفاض في هرمون الغدة الدرقية وهرمون الجار درقية وهرمونات الغدة النخامية
مشاكل في القلب كتضخم عضلة القلب مع هبوط في القلب وعدم أنتظام في دقات القلب .
احتمال أصابه المريض بالأمراض المعدية نتيجة لنقل الدم بشكل مستمر و لكن قلة تلك المخاطر بعد قيام جميع مراكز التبرع بالدم بفحص إذا ما كان دم المتبرع بدم مصاب بالإيدز او فيروسات التهاب الكبد الوبائي.
الثلاسيميا مرض وراثي ينتقل من الأبوين الحملين للمرض إلى بعض أطفالهم.ولكي تفهم الجزء الوراثي لهذا المرض عليك معرفة بعض الأمور المتعلقة بتصنيع الهيموجلوبين وبعض الأساسيات في علم الوراثة.ولكي تفهم هذا الجانب العلمي أنصحك بالرجوع إلى صفحة أساسيات الوراثة لمعرفة ما هي الخلية والنواة و الكروموسوم والمورث .وسوف أقوم بشرح مختصر عن هذا الكلمات في سياق كلامي عن الهيموجلوبين والمورثات في هذه الصفحة.
تصنيع الهيموجلوبين:
يصنع الهيموجلوبين داخل خلايا الدم الحمراء والتي هي الأخرى تصّنع في نخاع العظام(مخ العظم).والهيموجلوبين عبارة عن ستة قطع مترابطة مع بعضها البعض.القطعة الأولى هي الحديد وهي تلتصق بالقطعة الثانية وهي مادة الهيم (Hem )وهاتين القطعتين محاطة بأربع قطع من البروتين المسمى بالجلوبين(Globin )اثنتان من هذا الجلوبين من نوع ألفا و اثنتان من نوع بيتا وفي هذه الحالة يسمى هذا الهيموجلوبين بهيموجلوبين ( أ ) أو (A ).
هذا الترابط العجيب بين الحديد و الهيم و الجلوبين هو الذي يجعله مادة ناقلة للأكسجين في الدم.
كل هذه القطع ما عدى مادة الحديد تصنع داخل الجسم البشري.وكل قطعة هي عبارة عن مادة بروتينية .وكل البروتينات في جسم الإنسان تصنع داخل الخلية.وبتحديد داخل نواة الخلية ،وبشكل أدق داخل الكروموسومات(الصبيغات) ،وبشكل اكثر دقة داخل الجين(المورث).أي أن مادة الهيم ومادة الجلوبين يصنعها عدة مورثات موجودة على الكروموسومات.ومادة الهيم يصنعها 6 مورثات موزعة على عدة كروموسومات،واي خلل في هذه المورثات يسبب مجموعة من الأمراض الوراثية ولكنها تختلف عن الثلاسيميا.مرض الثلاسيميا يحدث نتيجة خلل (طفرة) في المورثات التي تصنع مادة الجلوبين.
المورثات و الجلوبين:
هناك نوعان أساسيان من مادة الجلوبين .الأولى تسمى ألفا جلوبين والأخرى تسمى البيتا جلوبين.وكما ذكرنا ففي كل هيموجلوبين قطعتان من الألفا وقطعان من البيتا جلوبين.ويصنع البيتا و الألفا جلوبين من مورثات موجود على الكروموسومات،كما هو الحال في مادة الهيم.
ويوجد مورثين اثنين لتصنيع مادة البيتا جلوبين واحدة موجودة على الكروموسوم 11 الذي ورثة الإنسان من أبوه والأخرى على النسخة الثانية من كروموسوم 11 الذي ورثة الإنسان من أمة..بينما يصنع مادة الألفا جلوبين من أربع مورثات،اثنتان موجودة على كروموسوم 16 الذي ورث من الأب والاثنين الآخرين على النسخة الثانية من كروموسوم 16الذي ورثة الإنسان من أمة التي جاءت من الأم.إذا حدث خلل أو عُطب (طفرة) في أحد مورثات الألفا جلوبين يصاب الإنسان بمرض ألفا ثلاسيميا، و إذا أصاب الخلل مورث من مورثات البيتا يصاب الإنسان بمرض البيتا ثلاسيميا.هذه بشكل مبسط العلاقة بين المورثات و الثلاسيميا،ولكن الموضوع اعقد من ما ذكرنا ،ولكي تعرف المزيد تابع القراءة!
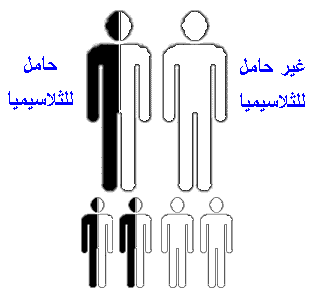
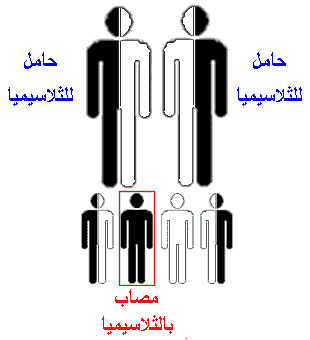
البيتا ثلاسيميا (Beta Thalassemia ) :خلاصة ما ذكرنا أن هناك أربع مورثات لتصنيع مادة الألفا جلوبين، و مورثين لتصنيع مادة البيتا جلوبين.واليك الآن بعض التفصيل في هذا الأمر و لنبدأ بالبيتا ثلاسيميا.لو أصابت الطفرة(أي العطب أو الخلل) نسخة واحدة من مورثات البيتا جلوبين فأن الخلية تستمر في إنتاج البيتا جلوبين وذلك نظرا لوجود مورث أخر سليم .ولكن هناك نقص في الكمية الإجمالية المنتجة من هذه المادة ولكن هذا النقص لا يسبب مرضا ولكن يسمى الشخص الذي يحمل مورث واحدا معطوبا من مورثات البيتا بالحامل للمرض،أي انه حامل لمرض البيتا ثلاسيميا ويمكن أن يتعرف على هذه الحالة بأجراء فحص للكريات الدم الحمراء والهيموجلوبين و فحص حركة الهيموجلوبين الإلكترونية ( Hemoglobin Electrophoresis)وهي متوفرة في الكثير من المستشفيات.أما لو حدثت الطفرة في كلا النسختان فان مجوع الكمية المصنعة من البيتا جلوبين تقل بشكل كبير فيصاب الشخص بمرض البيتا ثلاسيميا وتكون شدة المرض على حسب شدة الطفرة فبعض الطفرات تسمح بإنتاج كمية معقولة من مادة البيتا جلوبين(ويسمى المرض في هذه الحالة بالبيتا ثلاسيميا الصغرى)وهناك طفرات شديدة تؤدي إلى نقص كبير وقد يكون كامل في مادة البيتا جلوبين (ويسمى المرض في هذه الحالة بالبيتا ثلاسيميا). نظرا لوجود أربع مورثات من الألفا جلوبين فأن الشخص تظهر عليه أعراض المرض على حسب عدد المورثات المعطوبة .فإذا أصيب مورث واحد فأن الشخص لا يصاب بأي مشكلة صحية(يسمى بالألفا ثلاسيميا الساكتة Silent alpha Thalassemia) وقد يصعب التعرف على هذه الحالة عند إجراء تحليل دم عادي(أي عدد الكريات الحمراء والهيموجلوبين )ولا يمكن الكشف علية إلا بعمل فحص لحركة الهيموجلوبين الإلكتروني في الأشهر الأولى من العمر أما بعد هذا السن فأنه يصعب اكتشاف هذا الأمر الا بالكشف على المورثات مباشرة،وهذه غير متوفرة في الكثير ن المراكز الطبية.بينما فحص حركة الهيموجلوبين الإلكترونية ( Hemoglobin Electrophoresis) متوفر في الكثير من المستشفيات.
الالفا ثلاسيميا ( Alpha thalassemia):
بينما يصنع مادة الألفا جلوبين من أربع مورثات،اثنتان موجودة على كروموسوم 16 الذي ورث من الأب والاثنين الآخرين على النسخة الثانية من كروموسوم 16الذي ورثة الإنسان من أمة التي جاءت من الأم.إذا حدث خلل أو عُطب (طفرة) في أحد مورثات الألفا جلوبين يصاب الإنسان بمرض ألفا ثلاسيميا.
ونظرا لوجود أربع مورثات من الألفا جلوبين فأن الشخص تظهر عليه أعراض المرض على حسب عدد المورثات المعطوبة .فإذا أصابت الطفرة مورث واحد فقط فأن الشخص لا يصاب بأي مشكلة صحية و تسمى هذه الحالة بالألفا ثلاسيميا الساكتة Silent alpha Thalassemia).وفي العادة لا يعرف الشخص انه حامل لمورث معطوب وقد يصعب التعرف على هذه الحالة حتى عند إجراء تحليل دم عادي(أي عدد الكريات الحمراء والهيموجلوبين )ولا يمكن الكشف علية إلا بعمل فحص لحركة الهيموجلوبين الإلكتروني في الأشهر الأولى من العمر أما بعد هذا السن فأنه يصعب اكتشاف هذا الأمر إلا بالكشف على المورثات مباشرة،وهذه غير متوفرة في الكثير من المراكز الطبية.أما إذا أصابت الطفرة مورثين اثنين من الأربعة فان هذه الحالة تسمى بالحامل للصفة الألفا ثلاسيميا (Alpha Thalassemia Trait) وهذا الشخص أيضا لا يصب بمشاكل في الصحة وليس علية خطورة .ولكن يمكن الاشتباه في أن الشخص لدية هذا النوع من الثلاسيميا عند إجراء تحليل عادي لكريات الدم الحمراء ،ففي هذه الحالة تكون كريات الدم الحمراء اصغر من الحجم المعتاد وكمية الهيموجلوبين أيضا اقل من المعتاد.ولكن هذه العلامات التي على كريات الدم الحمراء أيضا تظهر عند بعض الأشخاص أو الأطفال المصابون بفقر الدم الناتج عن نقص تناول الحديد.لذلك يقوم الأطباء بقياس كمية الحديد في الدم فإذا كانت طبيعية فأن الاشتباه أن هذه الحالة ناتجة عن الألفا ثلاسيميا .أما إذا كان هناك نقص في الحديد فيعالج الأمر بإعطاء الحديد ثم يعاد التحليل .و إذا زال نقص الحديد واستمر صغر كريات الدم الحمراء فان هذا يعني أن الشخص كان عنده نقص في الحديد مصحوب بألفا ثلاسيميا.يكثر الاشتباه بين فقر الدم الناتج عن نقص الحديد و ألفا ثلاسيميا لذلك على العائلة تنبيه الطبيب على أن بعض أفراد العائلة لديهم ألفا ثلاسيميا لكي لا يعطى الطفل أو البالغ(كامرأة الحامل) جرعات من الحديد من دون أن يكون هناك ضرورة لهذا الأمر لأن ارتفاع كمية الحديد في الجسم مضر.أما إذا أصاب العطب(الطفرة)ثلاث مورثات من الأربعة فان الشخص يصاب بمرض يسمى بمرض الهيموجلوبين اتش (Hemoglobin H disease).وعند ولادة الطفل المصاب يكون هناك نقص في كمية الهيموجلوبين في كريات دمهم الحمراء مع وجود هيموجلوبين غير طبيعي يسمى بهيموجلوبين بارت وهو ناتج عن اتحاد أربع قطع من جلوبين جاما .وبعد مضي بعض الأشهر(أي خلال الفترة التي تقل فيها كمية الجاما جلوبين في جسم الإنسان) يصاب الطفل بفقر دم من النوع المتوسط فيتراوح مستوى الهيموجلوبين حوالي 8-10 غرامات لكل 100 ملليتر ويكون لدية تضخم في الطحال مع يرقان . بشكل عام لا يحتاج الأطفال لنقل الدم، أما البالغين فقد يحتاجون لنقل كميات من الدم.أما إذا أصاب العطب(الطفرة)جميع المورثات الأربعة يصاب الجنين باستسقاء شديد في جميع الجسم نتيجة لوجود فقر دم حاد مع فشل في القلب كنتيجة ثانوية لنقص الهيموجلوبين في الدم.ويموت الجنين إذا لم ينقل له جم في أول الحمل،و إذا ولد الطفل فانه يحتاج لنقل دم بشكل دوري نتيجة لوجود فقر دم شديد إلى متوسط الشدة.